Introduction
This document describes the usage of a .par parameter (input) file for MPI-AMRVAC. For a list of command line options, see Command line parameters.
Parameters are grouped in namelists according to their functionalities. The namelists have physics-independent class and physics-dependent class. The physics-independent class includes:
- Filelist Name and type of files to save (or read)
- Savelist When to save data
- Stoplist When to stop the simulation
- Methodlist Which numerical methods to use (e.g., flux scheme, time stepper, time integrator, limiter)
- Boundlist Boundary conditions
- meshlist Mesh-related settings (e.g. domain size, refinement)
- Paramlist Time-step parameters
The default parameter values in these namelists are set in src/amrvacio/mod_input_output.t, look at the subroutine read_par_files for details.
The physics-dependent namelists include:
- rho list (see also
mod_rho_phys) - nonlinear list (see also
mod_nonlinear_phys) - HD list (see also
mod_hd_phys) - MHD list (see also
mod_mhd_phys)
Further namelist are used to control optional modules. Most of these lists are not documented here, but the parameters are described in the corresponding modules:
rc_list(radiative cooling, seemod_radiative_cooling)tc_list(thermal conduction, seemod_thermal_conduction)dust_list(dust, seemod_dust)vc_list(viscosity, seemod_viscosity)grav_list(gravity, seemod_gravity)mf_list(magnetofriction, seemod_mf_phys)
An example for a namelist
The parameter file consists of a sequence of namelists, which look like this:
&listname var_a = value ! A comment var_b = value array = value1, value2, ... array(1,1) = value ! Repeat value 5 times in array array = 5*value ... /
If you do not define a variable the default value is used.
The Fortran 90 standard for logical variable values is either T and F or .true. and .false., but some compilers accept only one of them. Text between namelists is ignored, but it may result in a run time error on some machines/compilers.
The following namelist examples contain all the possible variables to set, choices are indicated by |. The first choice is the default value. Only the parameters different from default need to be set. Names that should be replaced are in capital letters. The ... indicates optional extra elements for arrays, or extra words in strings.
Physics-independent Namelists
Filelist
&filelist <variable definitions, see below> /
| name | type | default | description |
|---|---|---|---|
| base_filename | string | 'data' | Base file name for simulation output, which will be followed by a 4-digit number |
| restart_from_file | string | - | Resume from the snapshot data with this file name. |
| typefilelog | string | 'default' | Use 'regression_test' to do regression test and use the value 'special' to enable user-defined log output |
| snapshotnext | integer | 0 | Start index for writing snapshots |
| slicenext | integer | 0 | Start index for writing slices |
| firstprocess | logical | F | If true, call initonegrid_usr upon restarting |
| reset_grid | logical | F | If true, rebuild the AMR grid upon restarting |
| convert | logical | F | If true and filenameini and snapshotini are given, convert snapshots to other file formats |
| convert_type | string | vtuBCCmpi | Which format to use when converting, options are: tecplot, tecplotCC, vtu, vtuCC, vtuB, vtuBCC, tecplotmpi, tecplotCCmpi, vtuBmpi, vtuBCCmpi, vtumpi, vtuCCmpi, pvtumpi, pvtuCCmpi, tecline, teclinempi, onegrid, EIvtiCCmpi, ESvtiCCmpi, SIvtiCCmpi, EIvtuCCmpi, ESvtuCCmpi, SIvtuCCmpi |
| slice_type | string | vtu | Which format to use when slicing, options are: csv, dat, vtu, vtuCC |
| collapse_type | string | vti | Which format to use when slicing, options are: csv, vti |
| autoconvert | logical | F | If true, already convert to output format during the run |
| sliceascii | logical | F | If true, enable ASCII output of Slice output |
| saveprim | logical | F | If true, convert from conservative to primitive variables in output |
| nwauxio | integer | 0 | Number of auxiliary variables that are only included in the output |
| w_convert_factor | double(1:nw) | 1.0 | Conversion factors for w variables |
| time_convert_factor | double | 1.0 | Conversion factor for time unit |
| length_convert_factor | double | 1.0 | Conversion factor for length unit |
level_io | integer | - | When doing a convert, generate a uniform grid at this level |
level_io_min | integer | 1 | Minimum grid level when doing a convert |
level_io_max | integer | nlevelshi | Maximum grid level when doing a convert |
| nocartesian | logical | F | If true, do not convert the output to a Cartesian coordinate system |
| w_write | logical(1:nw) | all true | VTK: Only write variables for which writew(iw) is true |
| writelevel | logical(1:nlevelshi) | all true | VTK: only write these levels |
| writespshift | double(1:ndim,1:2) | all zero | clip off this relative amount of the domain at the lower and upper side in each dimension |
The log file
By default, the logfile contains one line with a string that is meant to identify the coordinate names, the conserved variables (wnames) and other entries, and then follows a sequence of lines containing numbers: i.e. a single line per requested output time, containing the integer timestep counter it, the time global_time, the time step to be used in the next time advance dt, the domain integrated value of each conserved variable (nw real numbers, which allows to check perfect conservation across the grid tree when the boundary conditions imply it), the percentage of the domain covered by each allowed grid level (given by refine_max_level real numbers between 0.0 and 1.0, with 1.0 indicating 100% coverage: when all numbers are summed, we get 1.0), and the number of grids per allowed grid level (hence, given by refine_max_level integers). The logfile is by default saved as an ASCII file. The order of saving snapshots, and regridding actions is fixed: regrid happens after the advance by one timestep, then regrid, then save the data. This has consequences for the optional variables beyond nwflux.
Further info on output and postprocessing
The code can be used to postprocess the MPI-AMRVAC .dat files (which are the only ones to be used for restarts) to some convenient data files for later visualisation purposes. Such conversion of a single .dat file at a time is to be done with the same executable (or at least one compiled on a possibly different machine), on a single processor (i.e. using mpirun -np 1 amrvac). Only selected output types can be converted in parallel, namely those whose name contains 'mpi' as part of the convert_type string. Currently, this includes the ASCII (binary) versions of vtumpi (vtuBmpi) with values located at cell corners and vtuCCmpi (vtuBCCmpi) with values at cell centers, and similarly for tecplot (tecplotmpi or tecplotCCmpi). In addition, pvtumpi (pvtuBmpi) and pvtuCCmpi (pvtuBCCmpi) result in parallel output with one *.vtu file for each processor and a header file from master processor.
In this conversion mode, the idea is to set restart_from_file together with convert=T. You can ask the code during conversion to change from conservative to primitive variable output by setting saveprim=T, and then the corresponding names for the primitive variables are automatically determined. It is also possible to perform the conversion step during run of the simulation with the switch autoconvert=T. Naturally, this leads to more computational overhead and IO, but using the pvtuB(CC)mpi filetype, this can be reduced to a minimum.
For simulations on non-cartesian grids (cylindrical or spherical), there is the option to output the non-cartesian cell coordinates and vector components, which then forces you typically to do the conversion to cell center cartesian grid coordinates and vector variables in your visualization session. By default (i.e. nocartesian=F), the convert module does the conversion from the orthogonal to locally cartesian coordinates and vector components for you. You can overrule this default behavior by setting nocartesian=T. (note: for tecplot format, the coordinate labels are then corrected in the converted file as well).
The only variable that then further matters is convert_type. For type 'tecplot', a corresponding base_filenamexxxx.plt file will be generated, which is an ASCII file that stores the cell corner locations and corner values for the conserved variables, to be handled with Tecplot. The 'onegrid' conversion type is just useful in 1D AMR runs, to generate a single block file (extension '.blk'). Also particular to 1D data, and for TecPlot purposes alone, is the 'tecline' option. This can also be done in parallel mode, where it is called 'teclinempi'.
For visualization using Paraview, the option to convert to ‘convert_type='vtu’` can be used. For both vtu and tecplot formats, there are also corresponding vtuCC and tecplotCC options, which store the data with the actually computed cell-centered values. For the vtu and tecplot formats on the other hand, the code tries to already do some interpolation from cell center to cell corner variables for you, but this may introduce some artificial effects in non-cartesian geometries. The vtuB and vtuBCC do the same as vtu(CC) but save the data in binary format.
It is even possible to temporarily add additionally computed auxiliary variables that are instantaneously computable from the data in the base_filenamexxxx.dat file to the converted snapshot. You should then provide the number of such extra variables in nwauxio (see also this page), and a corresponding definition for how to compute them from the available nw variables in the associated subroutine usr_special_convert whose default interface is provided in the mod_usr_methods.t module. You can there compute variables that are not in your simulation or data file, and store them in the extra slots nw+1:nw+nwauxio of the w variable. For consistency, you should also then add meaningfull names to a string to identify the auxiliary variables, this has to be done in the associated subroutine usr_add_aux_names.
The output values are normally given in code units, i.e. in the dimensionless values used throughout the computation (in the initial condition, we always adhere to the good practice of choosing an appropriate unit of length, time, mass and expressing everything in dimensionless fashion). One can, in the convert stage only, ask to multiply the values by their respective dimensional unit value. time_convert_factor should then be the unit for time, while the array w_convert_factor for w variables and length_convert_factor for length. The corresponding values for conservative entries are computed in convert.t when saveprim=F. See for details of their use the convert.t module.
Note that different formats for postprocess data conversion can be added in the convert.t subroutine. See convert for details.
The VTK-based formats allow for saving only a part of the nw variables, by setting the logical array w_write. The same is true for selecting levels by using the array writelevel. Finally, you can clip away part of the domain, for output in a selected region. This is done by filling the array writespshift. That array should use (consistent) double precision values (between 0 and 1) that specify the percentage of the total domain to be clipped away from the domain boundary at the minimum side, and from the maximum side, respectively, and this for all dimensions. The array has thus a dimensionality _(1:ndim,1:2)_, with the first entry specifying the dimension, and the second whether you clip for minimum (1) and maximum (2) sides, respectively. Note that in the end, only the grids that are fully contained within the clipped region are then converted and stored in the output.
The switches level_io, level_io_min and level_io_max are there to restrict the AMR levels of the output file at the convert stage. These switches do not work with autoconvert. E.g. setting level_io=3 will coarsen/refine the data to level 3 everywhere, resulting in a uniform grid. level_io_min limits the minimum level for output by refining levels below level_io_min until level_io_min is reached. Correspondingly, level_io_max limits the maximum level of the output file. This can be useful to visualize large datasets.
The convert types EIvtiCCmpi, ESvtiCCmpi, SIvtiCCmpi, EIvtuCCmpi, ESvtuCCmpi and SIvtuCCmpi are added for synthesize EUV images, EUV spectra and SXR images, where EI represents EUV image, ES represents EUV spectra and SI represents SXR image. The corresponding parameters , e.g. line of sight direction, for synthesizing emissions should be provided in the Synthetic EUV/SXR/whitelight emission when the convert types are activited.
Savelist
Example:
&savelist
itsave(1,1)=0
itsave(1,2)=0
dtsave_log=0.01d0
dtsave_dat=0.1d0
dtsave_slice=0.05d0
dtsave_collapsed=0.05d0
/
| name | type | default | description |
|---|---|---|---|
ditsave_log | integer | biginteger | Repeatedly save information in a log file when ditsave_log time steps have passed |
dtsave_dat | double | bigdouble | Repeatedly save dat files when dtsave_dat simulation time has passed |
itsave(SAVEINDEX,FILEINDEX) | integer | biginteger | Save on these time steps |
tsave(SAVEINDEX,FILEINDEX) | double | bigdouble | Save on these times |
nslices | integer | 0 | Number of slices |
slicedir(INTEGER) | integer | - | Slice direction, see Slice output |
slicecoord(INTEGER) | double | - | Slice coordinate, see Slice output |
collapse(INTEGER) | logical | F | See Looking at line-integrated quantities |
collapseLevel | integer | 1 | See Looking at line-integrated quantities |
Here FILEINDEX has the following meaning:
| index | meaning |
|---|---|
| 1 | Log output |
| 2 | Normal output |
| 3 | Slice output, see Slice output |
| 4 | Collapsed output, see Looking at line-integrated quantities |
| 5 | Call user custom analysis subroutine, see Writing a custom analysis subroutine |
One may want to save snapshots more frequently at the beginning of the simulation. E.g. tsave(1,2)=0.1 tsave(2,2)=0.25 tsave(3,2)=0.5 dtsave_dat=0.5 could be used to save snapshots at times 0.1, 0.25, 0.5, 1, 1.5, ... etc.
If no save condition is given for a file you get a warning, but the final output is always saved after the stop condition has been fulfilled. If itsave(1,2)=0 is set, the initial state is saved before advancing.
Stoplist
&stoplist
it_max =INTEGER
time_max =DOUBLE
wall_time_max =DOUBLE
dtmin =DOUBLE
it_init =INTEGER
time_init =DOUBLE
reset_time =F | T
reset_it =F | T
final_dt_reduction=T | F
/
| name | type | default | description |
|---|---|---|---|
it_max | integer | biginteger | a run ends when the number of time steps reaches it |
time_max | double | bigdouble | a run ends when the physical time reaches it |
wall_time_max | double | bigdouble | a run ends when the wall time reaches it (in hours) |
dtmin | double | bigdouble | a run stops when time step falls smaller than it |
it_init | integer | 0 | set the initial number of time steps when start a new run |
time_init | double | 0 | set the initial time when start a new run |
reset_time | logical | F | when restart from a previous run, reset the time to the initial one if it is T |
reset_it | logical | F | when restart from a previous run, reset the number of time steps to the initial one if it is T |
final_dt_reduction | logical | T | forces the last dt to comply with time_max limit |
You may use an upper limit it_max for the number of timesteps, and/or the physical time time_max, and/or the wall time 'wall_time_max' to end a run. The wall time means wall clock time recording real-world time.
Numerical or physical instabilities may produce huge changes or very small time steps depending on the way dt is determined. These breakdowns can be controlled by either setting a lower limit dtmin for the physical time step, which is useful when dt is determined from the courantpar parameter. If a run stops due to dt < dtmin, a warning message is printed.
You have to specify at least one of time_max, it_max. AMRVAC stops execution when any of the limits are exceeded. The initial time value time_init and integer time step counter it_init values, which are zero by default, can be specified here. However, when a restart is performed from a previous .dat file, the values in that file will be used unless you reset them to their initial values by setting reset_time=T. If you want only to reset the iteration count without changing time, set reset_it=T.
Methodlist
&methodlist time_stepper='twostep' | 'onestep' | 'threestep' | 'fourstep' | 'fivestep' time_integrator= choices depends on time_stepper flux_scheme=nlevelshi strings from: 'hll'|'hllc'|'hlld','hllcd'|'tvdlf'|'tvdmu'|'tvd'|'cd'|'fd'|'source'|'nul' limiter= nlevelshi strings from: 'minmod' | 'woodward' | 'superbee' | 'vanleer' | 'albada' | 'ppm' | 'mcbeta' | 'koren' | 'cada' | 'cada3' | 'mp5' | 'schmid1' | 'schimid2' | 'venk' | 'weno3' | 'wenoyc3' | 'weno5' | 'weno5nm'| 'wenoz5' | 'wenoz5nm' | 'wenozp5' | 'wenozp5nm' | 'weno5cu6' | 'teno5ad' | 'weno7' | 'mpweno7' gradient_limiter= nlevelshi strings from: 'minmod' | 'woodward' | 'superbee' | 'vanleer' | 'albada' | 'ppm' | 'mcbeta' | 'koren' | 'cada' | 'cada3' loglimit= nw logicals, all false by default flatsh = F | T flatcd = F | T mcbeta= DOUBLE typeentropy= 'nul'|'powell'|'harten'|'ratio'|'yee' entropycoef= DOUBLE, DOUBLE, DOUBLE, .... typetvd= 'roe' | 'yee' | 'harten' | 'sweby' typeaverage='default' | 'roe' | 'arithmetic' typeboundspeed= 'Einfeldt' | 'cmaxmean' | 'cmaxleftright' tvdlfeps = DOUBLE flathllc= F | T nxdiffusehllc = INTEGER source_split_usr= F | T typesourcesplit= 'sfs' | 'sf' | 'ssfss' | 'ssf' dimsplit= F | T typedimsplit= 'default' | 'xyyx'| 'xy' small_density= DOUBLE small_pressure= DOUBLE small_temperature= DOUBLE small_values_method='error' | 'replace' | 'average' small_values_daverage=1 check_small_values= F | T fix_small_values= F | T trace_small_values= F | T small_values_fix_iw= LOGICAL, LOGICAL, LOGICAL, ... typegrad = 'central' | 'limited' typediv = 'central' | 'limited' typecurl = 'central' | 'Gaussbased' | 'Stokesbased' /
time_stepper, time_integrator, flux_scheme
The time_stepper variable determines the time integration procedure. The default procedure is a second order predictor-corrector type 'twostep' scheme (suitable for TVDLF, TVD-MUSCL schemes), and a simple 'onestep' algorithm for the temporally second order TVD method, or the first order TVDLF1, TVDMU1, TVD1 schemes. It is not possible to mix different step size methods across the AMR grid levels. The temporally first order but spatially second order TVD1 algorithm is best suited for steady state calculations as a 'onestep' scheme. The TVDLF and TVD-MUSCL schemes can be forced to be first order, and linear in the time step, which is good for getting a steady state, by setting ‘time_stepper='onestep’`.
There is also a fourth order Runge-Kutta type method, when ‘time_stepper='fourstep’` and one sets ‘time_integrator='rk4’`. It can be used with dimsplit=.true.. These higher order time integration methods can be most useful in conjunction with higher order spatial discretizations. See also discretization and time_discretization.
The array flux_scheme defines a scheme to calculate the flux at cell interfaces using the chosen method (like hll based approximate Riemann solver) per activated grid level (and on each level, all variables use the same discretization). In total, nlevelshi methods must be specified, by default nlevelshi=20 and these are then all set by flux_scheme=20*'tvdlf'. Different discretizations can be mixed across the refine_max_level activated grid levels (but the same stepping scheme must apply for all of the schemes).
Setting for a certain level the flux_scheme to 'nul' implies doing no advance at all, and 'source' merely adds sources. These latter two values must be used with care, obviously, and are only useful for testing source terms or to save computations when fluxes are known to be zero.
Limiter type
For the TVDLF and TVD-MUSCL methods different limiter functions can be defined for the limited linear reconstructions from cell-center to cell-edge variables, and for the TVD method, for the characteristic variables. The default limiter is the most diffusive ‘limiter=nlevelshi*'minmod’` limiter (minmod for all levels), but one can also use ‘limiter=nlevelshi*'woodward’`, or use different limiters per level. Click Slope limiters to see detailed description of all available limiters.
The gradient_limiter is the selection of a limiter to be used in computing gradients (or divergence of vector) when the typegrad=limited (or typediv=limited) is selected. It is thus only used in the gradientS (divvectorS) subroutines in geometry.t and has effect for the magnetohydrodynamics (MHD) modules.
When having a gravitational stratification, one might benefit from performing linear reconstruction on the primitive variables log10(rho) and/or log10(p). This can be done by setting the corresponding loglimit(iw)=T with iw the label of the corresponding component in the w array (for density, this is thus iw=1).
When using PPM as a limiter, minor differences can be obtained using the switches flatcd and flatsh. These are meant to minimize potential ripples around contact discontuinities (flatcd) or shocks (flatsh), but one should first try without these flattenings (default behavior). PPM is actually only used in a quadratic reconstruction from center to edge, requires the use of a larger stencil (nghostcells=3), and can be used either in the methods (by setting limiter) or in the gradientS/divvectorS routines (when typegrad or typediv is limited, and gradient_limiter is ppm) in geometry.t. If flatcd=T or flatsh=T to flattern oscillations at contact discontuinities or shocks, it needs 1 more ghost cell, e.g., nghostcells=4.
Typeentropy, entropycoef
For Riemann solver based methods, such as TVD and TVD-MUSCL (but not TVDLF), an entropyfix may be applied to avoid unphysical solutions. The fix is applied to the characteristic variables. The default entropy fix is ‘'nul’`, i.e. no entropy fix. When an expansion shock is formed, the entropy fix should be applied to the non-degenerate characteristic waves, i.e. waves that can form shocks (sound waves, fast and slow magnetosonic waves). The most powerful entropy fix is called 'powell'. In practice, one may apply an entropy fix to all characteristic waves, usually the slight extra diffusion makes the schemes more robust. For Yee entropyfix the minimum characteristic speed (normalized by dt/dx) can be set for each characteristic wave using the entropycoef array.
Different TVD variants
Both tvd and tvdlf have a few variants, these can be set in the strings typetvd and typeboundspeed, with defaults 'roe' and 'Einfeldt', respectively. The default ‘typetvd='roe’‘ is the fastest of the four upwind types. For the TVDLF, the 'cmaxmean’ merely means whether the maximal physical propagation speed is determined as the maximum speed for the mean state based on averaging left centered and right centered states, or by taking the Roe average eigenvalues for the left and right nonlinear waves proposed by Einfeldt (Einfeldt 1988 J. Numer. Anal.). In the TVDLF flux, the diffuse flux part has a coefficient tvdlfeps which is 1 by default. For steady- state computations, one may gain in shock sharpness by reducing this factor to a positive value smaller than 1.
Only for the adiabatic hydro module, the option to select an arithmetic, or a roe average is available for use in the roe solver. This is set by the typeaverage.
Just like in the TVDLF method, the slightly more involved HLL method has a diffuse flux part with coefficient tvdlfeps which is 1 by default. For steady-state computations, one may gain in shock sharpness by reducing this factor to a positive value smaller than 1.
When using the HLLC scheme variants, for HD, MHD there is an optional additional flattening in case the characteristic speed at the contact is near zero. This is activated by setting flathllc=T (its default is false). One can also solve some potential noise problems in the HLLC by switching to the HLLCD variant, a kind of mix between HLLC and TVDLF. The TVDLF is then used in a user-controlled region around a point where there is a sign change in flux, whose width is set by nxdiffusehllc (an integer which is 0 by default).
Dimensional splitting
Special sources, if any, can be added in a split or unsplit way according to the logical variables source_split_usr The default value is false meaning these sources are added in an unsplit way by default. The split sources are added according to typesourcesplit. The meaning of the different options for typesourcesplit is described in discretization. Under default settings, we use unsplit sources only, and if one reverts to split sources, ‘typesourcesplit='sfs’`.
In multidimensional calculations dimensional splitting can be used by setting dimsplit=T, with an alternating order of the sweeps ‘typedimsplit='xyyx’by default. It is best to use dimsplit=F`, the default value, but the TVD method needs a dimensionally split strategy. The limitations on using dimensionally unsplit methods are described in methods.
Positivity fixes
Negative pressure or density caused by the numerical approximations can make the code crash. For HD and MHD modules this can be monitored or even cured by the handle_small_values subroutines in each substep of iteration. The control parameters small_density, small_pressure, small_temperature play a role here: they can be set to small positive values but not negative values, while their default is 0. If small_temperature is positive, small_pressure is overwritten by the product of small_rho and small_temperature. If check_small_values is set to .true., an additional check for small values will be triggered in phys_get_pthermal. NOTE: If small values are detected then a crash will occur regardless of whether any other positivity fixes are enabled. If fix_small_values is set to .true., small values are actually treated or fixed in subroutines such as phys_to_primitive, phys_to_conserved and source terms like resistive terms in MHD.
The actual treatment is determined by the small_values_method parameter: Its default value 'error' causes a full stop in the handle_small_values subroutine in the physics modules. In this way, you can use it for debugging purposes, to spot from where the actual negative pressure and unphysical value gets introduced during which call. If the compilation is in debug mode and trace_small_value=T, then the error message shows the path of execution as number of lines of subroutines so that you know when the small values are encountered in the execution sqeuence. If it is somehow unavoidable in your simulations, then you may rerun with a recovery process turned on as follows. When small_values_method='replace', the parameters small_pressure, small_density are used to replace any unphysical value and set momentum to be 0, as encoded in mod_small_values.t. When you select small_values_method='average', any unphysical value is replaced by averaging from a user-controlled environment about the faulty cells. The width of this environment in cells is set by the integer small_values_daverage. The parameter small_values_fix_iw is an array, containing a logical for each variable. It has a length nw. It can be used to make sure that a certain variable is not adjusted by the small_values_method. The default is true for all the variables. If the user sets it to false for a given variable, that variable will not be adjusted by the small_values_method. The first elements are the logicals for the conservative variables in their standard order. As an example: if the user would like to adjust the pressure and density but not the momentum in a 2D MHD simulation, then small_values_fix_iw has to be set to T, F, F, T, T, T.
Special process
User controlled special process can be added to each iteration. Subroutine usr_process_grid can be registered in mod_usr.t to process for each grid. Subroutine usr_process_global can be registered in mod_usr.t to do global process. For example, you can do computations of non-local auxiliary variables (like the divergence of some vector fields, time integrals etc).
The typegrad can be selected to switch from simple centered differencing on the cell center values, to limited reconstruction followed by differencing when computing gradients. They call either of gradient ('central') or gradientS ('limited') subroutines that are themselves found in the geometry.t module. Similarly, a switch for the divergence of a vector is the typediv switch. When the 'limited' variant is used, one must set the corresponding gradient_limiter array to select a limiter (per level).
Boundlist
&boundlist nghostcells= INTEGER typeboundary_min^D= 'cont'|'symm'|'asymm'|'periodic'|'special'|'noinflow' typeboundary_max^D= 'cont'|'symm'|'asymm'|'periodic'|'special'|'noinflow' internalboundary = F | T ghost_copy= F | T /
The boundary types have to be defined for each conserved variable, except for psi (in GLM-MHD) and tracer fluids, at each physical edge of the grid. For 2D hydrodynamics they are: rho,m1,m2,e at the left boundary (typeboundary_min1); rho,m1,m2,e at the right (typeboundary_max1); rho,m1,m2,e at the bottom (typeboundary_min2); rho,m1,m2,e at the top boundary (typeboundary_max2). Boundary types of psi (in GLM-MHD) and tracer fluids are automatically set to be the same as boundary type of density by default. Boundary types of dust density and dust momentum still must be set manually by user, if dust module is activated. The general subroutine devoted to the treatment of boundary conditions (either customized by the user or not, internal or external to the simulation space, polar or not) is get_bc and the main files concerned are mod_ghostcells_update.t and boundary_conditions.t. Since the pre-defined boundary conditions are applied to the conserved variables, it does not guarantee the continuity of the fluxes (i.e. the terms associated to the velocity within the divergences in the fundamental equations under their conservative form, see equations.md) and can prevent the fluid from reaching a steady state. For instance, a conserved total specific intern energy (i.e. intern plus kinetic energy) e does not result, in general, in a conserved flux of the variable carried by the velocity field i.e. e+P, where P is the pressure. Without a source term, it means that the time variation of e can not cancel out.
Instead of manually specifying one by one the boundary conditions, the user can write 8*'X' to replace _'X'_ 8 times in a row for instance. Beware, it is simply a syntax substitution rule which does not tell anything about the number of variables nor the number of dimensions. To improve readability, users are invited to highlight this underlying structure in the instructions. For instance, in a two dimensional hydrodynamical simulation space (ndim=2) with the mass density, three components of the velocity field (ndir=3) and an energy equation, if the bottom boundary is a plane of symmetry, the upper boundary is opened and the lateral boundaries are periodic, we would write :
&boundlist typeboundary_min1= 5*'periodic' typeboundary_max1= 5*'periodic' typeboundary_min2= 'symm','symm','asymm','symm','symm' typeboundary_max2= 5*'cont' /
The default number of ghost cell layers used to surround the grid (and in fact each grid at each level and location) is set by default to nghostcells=2 and automatically increased if larger stencil is needed for high-order reconstructions. For example, when limiter=mp5 it takes 3, and limiter=ppm makes it 4.
The default boundary type is cont for all variables and edges, it means that the gradient (of the conservative variables) is kept zero by copying the variable values from the edge of the mesh into the ghost cells.
Other predefined types are the symm and asymm types, which are mostly used for reflective boundaries, or at symmetry axes of the domain (the polar or equatorial axis, e.g.). One then typically makes the momentum orthogonal to the given boundary antisymmetric (asymm), the rest of the variables symm. These boundary types can also be used to represent a perfectly conducting wall (the orthogonal component of the magnetic field should be antisymmetric, the transverse component symmetric) or the physical symmetry of the physical problem. More generally, true (a.k.a. polar) vectors (resp. pseudovectors, a.k.a. axial vectors) such as the ones associated to a velocity (resp. magnetic) field, transform such as the normal component (resp. the tangential components) is antisymmetric while the tangential components (resp. the normal component) are symmetric with respect to a plane of symmetry of causes (distribution of mass, of currents, of charges, etc). And vice versa for a plane of antisymmetry.
If the pole is adjacent to the simulation space (i.e. if the simulation extends down to a distance to the axis of 0 in cylindrical coordinates and if the simulation extends down to a colatitude of 0 or up to a colatitude of pi in spherical coordinates), symm and/or asymm boundary conditions must necessarily be specified. Scalar quantities are always symmetric. For a vector, whatever its nature (true vector or pseudovector), the boundary condition to choose for a given component depends on the behaviour of the axis vector associated to this component when a rotation of pi is performed within the plane orthogonal to the pole. In cylindrical coordinates, it means that the radial and vertical components should be symmetric while the orthoradial component is antisymmetric. In spherical coordinates, it means that the radial component is symmetric while the two remaining components are antisymmetric. For 3D cylindrical and spherical grid computations, the singular polar axis is trivially handled using a so-called pi-periodic boundary treatment, where periodicity across the pole comes from the grid cell diagonally across the pole, i.e. displaced over pi instead of 2 pi. These are automatically recognized from the coordinate setting, and the corresponding range in angle phi must span 2 pi for cylindrical, and theta must then start at zero (to include the north pole) and/or end at pi (for the south pole) for spherical grids. The user just needs to set the typeboundary as typeboundary_min2=8*'pole' and/or typeboundary_max2=8*'pole' in 3D spherical coordinates or typeboundary_min1=8*'pole' in cylindrical/polar coordinates, or use symmetry boundary (using symm and asymm combinations).
The case of periodic boundaries can be handled with setting 'periodic' for all variables at both boundaries that make up a periodic pair. Hence triple periodic in 3D MHD where 8 variables are at play means setting
typeboundary_min1=8*'periodic' typeboundary_max1=8*'periodic' typeboundary_min2=8*'periodic' typeboundary_max2=8*'periodic' typeboundary_min3=8*'periodic' typeboundary_max3=8*'periodic'
The possibility exists to put a boundary condition mimicking zero across the computational boundary, by selecting typeboundary='noinflow' for the momentum vector components of your particular application. This is in principle only relevant for the momentum component locally perpendicular to the boundary (for others a continuous extrapolation is done). The noinflow extrapolates values that are outwardly moving continuously, while clipping all values that are inwardly advecting momentum to zero.
The special type is to be used for setting fixed values, or any time dependent or other more complicated boundary conditions, and results in a call to the usr_special_bc subroutine which has to be provided by the user in the mod_usr.t module. The variables with special boundary type are updated last within a given boundary region, thus the subroutine may use the updated values of the other variables. The order of the variables is fixed by the equation module chosen, i.e. rho m1 m2 m3 e b1 b2 b3 for 3D MHD. It is suggested to set all typeboundary entries for a certain boundary region to special to consistently fill the boundary info for all variables in a user-defined manner.
Internal boundaries can be used to overwrite the domain variables with specified values through usr_internal_bc subroutine. This is activated with the switch internalboundary=T. Internally, these are assigned before the ghost-cells and external boundaries are applied (in subroutine get_bc).
The ghost_copy=F implies the use of limited linear reconstructions in the filling of ghost cells for internal boundaries that exist due to the AMR hierarchy. If ghost_copy=T, a first order 'copy' is used 'unlimit'. To retain second order accuracy, the default set is needed.
meshlist
&meshlist
refine_max_level= INTEGER
domain_nx1= INTEGER
domain_nx2= INTEGER
domain_nx3= INTEGER
block_nx1= INTEGER
block_nx2= INTEGER
block_nx3= INTEGER
xprobmin1= DOUBLE
xprobmax1= DOUBLE
xprobmin2= DOUBLE
xprobmax2= DOUBLE
xprobmin3= DOUBLE
xprobmax3= DOUBLE
refine_criterion= INTEGER
nbufferx1= INTEGER
nbufferx2= INTEGER
nbufferx3= INTEGER
max_blocks= INTEGER
amr_wavefilter= nlevelshi DOUBLE values
refine_threshold= nlevelshi DOUBLE values
derefine_ratio= nlevelshi DOUBLE values
w_refine_weight= DOUBLE array nw+1 values that must sum up to 1.0d0
logflag= nw logical values, all F by default
iprob= INTEGER
prolongprimitive= F | T
coarsenprimitive= F | T
tfixgrid= DOUBLE
itfixgrid= INTEGER
ditregrid= INTEGER
stretch_dim= ndim STRING values ('uni','symm','none')
stretch_uncentered = F | T
qstretch_baselevel= DOUBLE
nstretchedblocks_baselevel= INTEGER
/
refine_max_level, max_blocks, domain_nx^D, block_nx^D, xprobmin^D, xprobmax^D
refine_max_level indicates the maximum number of grid levels that can be used during the simulation, including the base grid level. It is an integer value which is maximally equal to the parameter nlevelshi and minimally equal to 1. The parameter nlevelshi=20 by default, a value set in mod_global_parameters.t, so that if more than 20 levels are to be used, one must change this value and recompile. Note that when refine_max_level>1, it is possible that during runtime, the highest grid level is temporarily lower than refine_max_level, and/or that the coarsest grid is at a higher level than the base level. The number of grid blocks in each processor has an upper limit defined by max_blocks which is 4000 by default and can be set to higher numbers if too many blocks in a processor are allocated.
The computational domain is set by specifying the minimal and maximal coordinate value per direction in the xprob^L settings. When cylindrical or spherical coordinates are selected, the angle ranges (for phi in the cylindrical case, and for both theta and phi in the spherical case) are to be given in 2 pi units.
The base grid resolution (i.e. on the coarsest level 1) is best specified by providing domain_nx^D, the number of grid cells per dimension, to cover the full computational domain set by the xprobmin^D and xprobmax^D. The resolution of each grid block is set by block_nx^D which exclude ghost cells at each side. The domain_nx^D must thus be a integer multiple of block_nx^D. The number of blocks on level 1 in a dimension, which does not have any pole or periodic boundary, must be larger than 1 for a rectanuglar mesh with more than one level because of the limitation from ghost-cell exchange at physical boundaries.
refine_criterion, nbufferx^D, amr_wavefilter
The code offers various choices for the error estimator used in automatically detecting regions that need refinement.
When refine_criterion=1, all refinement will only be based on the user- defined criteria to be coded up in subroutine specialrefine_grid.
When refine_criterion=3, the default value, errors are estimated using current t_n values and their gradients following Lohner prescription. In this scheme, the amr_wavefilter coefficient can be adjusted from its default value 0.01d0. You can set different values for the wavefilter coefficient per grid level. This error estimator is computationally efficient, and has shown similar accuracy to the Richardson approach on a variety of test problems. When refine_criterion=3, the original Lohner method is used.
A call to the user defined subroutine usr_refine_grid follows the error estimator, making it possible to use this routine for augmented user-controlled refinement, or even derefinement.
Depending on the error estimator used, it is needed or advisable to additionally provide a buffer zone of a certain number of grid cells in width, which will surround cells that are flagged for refinement by any other means. It will thus trigger more finer grids. nbufferx^D=2 is usually sufficient. It can never be greater than the block size. For Lohner scheme, the buffer can actually be turned off completely by setting nbufferx^D=0 which is default value.
w_refine_weight, logflag, refine_threshold, derefine_ratio
In all error estimators mentioned above (except the refine_criterion=1 case), the comparison or evaluation is done only with a user-selected (sub)set of the conserved variables. The nw variables (which may include auxiliary variables) can be used for error estimation, by setting corresponding weights in the array w_refine_weight, which by default has the first element (corresponds to density) as 1.d0 and rest elements as 0.d0. The weights w_refine_weight(:) must be positive values between 0.0d0 and 1.0d0, and must add to unity.
The Lohner error estimation (refine_criterion=3) may also decide to use differences in the log10(rho), and this is then done by setting the logflag(1)=T. This can be done per selected variables involved in the estimation, but obviously only works for those that remain positive throughout.
In the comparison involving the above selected variables, when the total error exceeds the value set by refine_threshold, the grid is triggered for refining. Reversely, if the error drops below derefine_ratio * refine_threshold, the grid is coarsened. The user must always set a (problem dependent) value for refine_threshold (below 1), while the default value for derefine_ratio=1.0d0/8.0d0 has shown to be a rather generally useful value. You can set threshold values that differ per refinement level.
When subroutine usr_refine_threshold is registered, user can use it to modify refine_threshold depending on location of interest. For example, increase refine_threshold near quiet boundaries to use coarser blocks there or decrease it to use finer blocks in focused regions.
iprob
As a possible integer switch for selecting multiple problem setups in the same executable code, the integer switch iprob is provided. It is meant to be used only in the user-written subroutines, for switching between e.g. multiple initial conditions for the same executable.
prolongprimitive, coarsenprimitive
It is possible to enforce the code to use primitive variables when coarsening grid information (coarsen), or filling new finer level grids (prolongation). They are then used instead of the conservative variables, which may not be a wise choice, but is perhaps better behaved with respect to positivity of pressure etc. This is activated seperately for prolongation by prolongprimitive=T, and for coarsen by coarsenprimitive=T.
The parameters tfixgrid, itfixgrid are available to fix the AMR hierarchical grid from a certain time onwards (tfixgrid) or iteration (the it- counter for the timesteps) onwards (itfixgrid). This may be handy for steady- state computations, or for those cases where you know that the initial conditions and physical setup is such that the AMR structure at t=0 will be optimal for all times.The parameter ditregrid is introduced to reconstruct the whole AMR grids once every ditregrid iteration(s) instead of regridding once in every iteration by default.
stretch_dim, stretch_uncentered, qstretch_baselevel, nstretchedblocks_baselevel
We allow stretching of the grid, in combination with any coordinate system (cartesian/polar/cylindrical/spherical) you choose. You activate grid stretching by setting stretch_dim(1:ndim), for example for the second dimension:
stretch_dim(2) = 'none' | 'uni' | 'symm'
- 'none' means don't stretch this dimension, which is the default.
- 'uni' means unidirectional stretching, where the grid cells change by a constant factor from cell to cell. The factor for the lowest refinement level can be set by setting
qstretch_baselevel=1.01(typical values are 1.01 to 1.05 or so, although any number larger than 1 is possible). - 'symm' means symmetric stretching, which is e.g. useful for setting up periodic domain problems or so. You then specify how many blocks you want to have unstretched (uniform) in the middle. E.g., you may have set up 8 blocks along a dimension at level 1, and then you can ask nstretchedblocks_baselevel=2,4,6 or 8.
Stretching can be useful for the radial coordinate in polar/spherical/cylindrical, or you can set the angle theta in 3D spherical to be stretched symmetrically, to leverage the CFL condition.
The parameter stretch_uncentered (default: true) controls whether mod_geometry.t routines such as divvector() take into account that a cell face is not between stretched cell-centers. However, this is not yet taken into account in the reconstruction and symm/asymm boundary conditions, which may lead to issues, which can sometimes be avoided by setting stretch_uncentered to false.
Paramlist
¶mlist dtpar= DOUBLE courantpar= DOUBLE typecourant= 'maxsum' | 'summax' | 'minimum' dtdiffpar= DOUBLE slowsteps= INTEGER /
dtpar, courantpar, typecourant, dtdiffpar, dtTCpar, slowsteps
If dtpar is positive, it sets the timestep dt, otherwise courantpar is used to limit the time step based on the Courant condition. The default is dtpar=-1. and courantpar=0.8.
For resistive MHD, the time step is also limited by the diffusion time: dt < dtdiffpar*dx^2/eta. The default is dtdiffpar=0.5. Further restrictions on the time step can be put in the usr_get_dt subroutine in the mod_usr.t. The library routines for viscosity and div B diffusive cleaning, all use the coefficient dtdiffpar in their stability conditions. The ‘typecourant='maxsum’` means that the time step limit for the CFL conditions takes the maximum over a summed contribution to the maximum physical propagation speed for all dimensions. The detailed formulae are found in setdt.t.
If the slowsteps parameter is set to a positive integer value greater than 1, then in the first slowsteps-1 time steps dt is further reduced according to the dt= dt * [ 1 - (1-step/slowsteps)**2 ] formula, where step=1..slowsteps-1. This reduction can help to avoid problems resulting from numerically unfavourable initial conditions, e.g. very sharp discontinuities. It is normally inactive with a default value -1.
Physics-dependent Namelists
rho list
&rho_list rho_v= ndim doubles for advection velocity /
nonlinear list
&nonlinear_list nonlinear_flux_type= INTEGER kdv_source_term= F | T /
HD list
&hd_list hd_energy= T | F hd_n_tracer= INTEGER hd_gamma= DOUBLE hd_adiab= DOUBLE hd_dust= F | T hd_thermal_conduction= F | T hd_radiative_cooling= F | T hd_gravity= F | T hd_viscosity= F | T hd_particles= F | T He_abundance= DOUBLE from 0 to 1 SI_unit= F | T /
MHD list
&mhd_list mhd_energy= T | F mhd_n_tracer= INTEGER mhd_gamma= DOUBLE mhd_adiab= DOUBLE mhd_eta= DOUBLE mhd_eta_hyper= DOUBLE mhd_etah= DOUBLE mhd_glm_alpha= 0.5d0 between 0 and 1 mhd_glm_extended= T | F mhd_magnetofriction= F | T mhd_thermal_conduction= F | T mhd_radiative_cooling= F | T mhd_Hall= F | T mhd_gravity= F | T mhd_viscosity= F | T mhd_particles= F | T mhd_4th_order= F | T mhd_internal_e= F | T mhd_hydrodynamic_e= F | T mhd_semirelativistic= F | T mhd_boris_simplification= F | T mhd_reduced_c = 6.d8 mhd_trac= F | T mhd_trac_type= INTEGER from 1 to 6 mhd_trac_mask= bigdouble mhd_trac_finegrid= INTEGER typedivbfix= 'linde'|'ct'|'glm'|'powel'|'lindejanhunen'|'lindepowel'|'lindeglm'|'multigrid'|'none' type_ct='uct_contact'|'uct_hll'|'average' source_split_divb= F | T boundary_divbfix= 2*ndim logicals, all false by default divbdiff= 0.8d0 between 0 and 2 typedivbdiff= 'all' | 'ind' clean_initial_divb= F | T divbwave= T | F B0field= F | T B0field_forcefree= T | F Bdip= DOUBLE Bquad= DOUBLE Boct= DOUBLE Busr= DOUBLE He_abundance= DOUBLE from 0 to 1 SI_unit= F | T /
Magnetic field divergence fixes
The upwind constrained transport methods typedivbfix='ct' by Gardiner and Stone in Journal of Computational Physics, 205, 509-539 (2005) type_ct='uct_contact' (default), or by Del Zanna, L., Zanotti, O., Bucciantini, N., & Londrillo, P. in Astronomy & Astrophysics , 473, 11 (2007) type_ct='uct_hll', using staggered grid for magnetic field, can preserve initial div B to round off errors. A simple non-upwinding version of ct is through averaging electric fields from neighbors type_ct='average'. And it only works with HLL, HLLC, and HLLD schemes in the current implementation. It works in Cartesian and non-Cartesian coordinates with or without grid stretching. It works with finite non-zero resistivity. Initial conditions and boundary conditions for magnetic field have to be given at corresponding cell faces instead, or vector potential is given at corresponding cell edges, see examples: tests/mhd/solar_atmosphere_2.5D. Note that when using AMR, the div B preserving prolongation for CT requires even number of ghost cell layers and odd number of ghost layers for some slope limiters, e.g. mp5, is added by one to become even.
In cell-center based magnetic fields, sources proportionate to the numerical monopole errors can be added, in a source-split way, to momemtum, energy, and induction equation (the 'powel' type), or to the induction equation alone (the 'janhunen' type) for cleaning divB errors. The divbwave switch is effective for the Riemann type solvers for multi-D MHD only. The default true value corresponds to Powell divergence wave which stabilizes the Riemann solver.
Another source term strategy for monopole error control is choose 'linde' type to do parabolic cleaning, i.e. add source terms which diffuse the local error at the maximal rate still compliant with the CFL limit on the time step. This is activated when divbdiff is set to a positive number, which should be less than 2, and again comes in various flavors depending on which equations receive source terms. The choice where only the induction equation gets modified, i.e. ‘typedivbdiff='ind’` can be used.
GLM-MHD mixed hyperbolic and parabolic dampening of the divB error using an additional scalar variable Psi. The algorithm of 'glm' is described by Dedner et al. as Equation (24) in Journal of Computational Physics 175, 645-673 (2002) doi:10.1006/jcph.2001.6961. The default one with parameter mhd_glm_extended=.true. is the extended GLM-MHD with divb related sources terms in momemtum and energy equation. The GLM-MHD method without the sources terms is used when mhd_glm_extended=.false.. The parameter mhd_glm_alpha ranging from 0 to 1.0 is the ratio of the diffusive to advective time scales for divb cleaning. You can choose 'lindejanhunen', 'lindepowel', or 'lindeglm' to use combined divb cleaning.
Projection scheme using multigrid Poisson solver by Teunissen and Keppens in Computer Physics Communications 245, 1068, (2019) can be chosen as 'multigrid' to remove div B part of B. If clean_initial_divb is set to T, the projection scheme is used to remove div B once after initial condition in unstretched Cartesian CT-MHD cases.
Magnetic field splitting strategy
For MHD, we implemented the possibility to use a splitting strategy following Tanaka, where a time-invariant background magnetic field is handled exactly, so that one solves for perturbed magnetic field components instead. This method works with HLLD, HLL and TVDLF flux and with all divb cleaning methods. The magnetic field splitting is activated by B0field=T, and the magnitude of the background magnetic field can be controlled using the parameters Bdip, Bquad, Boct, Busr. The first three are pre-implemented formulae for a dipole, quadrupole and octupole field in spherical coordinates only (the parameters set the strength of the dipole, quadrupole and octupole field). This is coded up in the module set_B0.t. This same module calls in addition the usr_set_B0 subroutine when Busr is non-zero, where it then should be used to quantify an additional time-independent field. This latter can be used for cartesian or cylindrical coordinates as well. User can possibly prescibe analytic current in usr_set_J0 subroutine to significantly increase accuracy. Choose B0field_forcefree=T when your background magnetic field is forcefree for better efficiency and accuracy.
The transition region adaptive conduction (TRAC) method
For solar atmosphere, simulations can sometimes suffer from the huge temperature gradient in the transition region. If the resolution is not enough (typically 1 km), the upward evaporation might be underestimated. The TRAC method could be used to correct the evaporation through artificially broadening the transition region. Set 'mhd_trac=T' to enable this function. 'mhd_trac_type=1' is the basic TRAC method for 1D simulation (see Johnston et al. 2019, 2020). When it is used for 2D or 3D simulations, the local cutoff temperature inside each block would be calculated separately. It is the fastest way in multi-D simulations, it is not accurate but basically, is physically correct. For multi-D uniform Cartesian grids, we prepared some other methods. 'mhd_trac_type=3' is the multi-D TRAC method based on the field line tracing module. For multi-D simulations, it should be the most accurate one, but might be very slow. Considering that this TRAC modificaiton will mostly affect the transition region, one can use 'mhd_trac_type=5' to add a mask to limit the region where the field lines are integrated. Give 'mhd_trac_mask' to set the maximum height of the mask, in your unit_length. 'mhd_trac_type=4' uses the block-based TRAC method for multi-D simulations, which should be faster than the second type. And 'mhd_trac_type=6' works in a similar way with the 4th type, by adding a mask on the block-based TRAC method. Give 'mhd_trac_finegrid' to set the distance between two adjacent traced field lines (in the unit of finest cell size). Note that when setting 'mhd_trac_type >=3', the direction of your gravity should follow y-dir (2D) or z-dir(3D).
'mhd_trac_type=2' is anthor TRAC method, which broadens the transition region according to a different criterion. Unlike Johnston TRAC method, this TRAC method has the advantage that all the calculation is done locally within the block. Thus, it could be used in either 1D (M)HD or multi-D MHD simulations, and is much faster than other multi-D methods.
Solve internal or hydrodynamic energy to avoid negative pressure
In extremely low beta plasma, internal energy or gas pressure easily goes to negative values when solving total energy equation, because numerical error of magnetic energy is comparable to the internal energy due to its extremely small fraction in the total energy. We have two methods to avoid this problem. In the first method, we solve internal energy equation instead of total energy equation by setting mhd_internal_e=T. This function is compatible with all finite volume and finite difference schemes we have, including HLL, HLLC, and HLLD, in which the Riemann flux of the internal energy is evaluted as the HLL flux in all intermediate states of the Riemann fan. In the second method, We solve hydrodynamic energy, i.e., internal and kinetic energy, instead of total energy with an additional source term of Lorentz force work, by setting mhd_hydrodynamic_e=T, which has better conservation than solving internal energy.
Solve semirelativistic MHD to tackle extreme Alfven speed
The Alfven speed in nonrelativistic MHD, defined as B/sqrt(mu rho), can be extremely large, even unphysically larger than speed of light in strong magnetic field and low density regions. Semirelativistic MHD equations (Gombosi et al. 2002 JCP 177, 176) as the nonrelativistic hydrodynamic limit of the relativistic MHD equations solve the problem and have the same steady-state solution as nonrelativistic MHD. By artificially lowering the speed of light, one can reduce the wave speeds allowing larger time steps thus faster solution in explicit numerical schemes. Set 'mhd_semirelativistic=T' and 'mhd_reduced_c' equals to a value smaller than light speed with physical unit to solve semirelativistic MHD. If setting 'mhd_hydrodynamic_e=T' or 'mhd_internal_e=T', the approximate split semirelativistic MHD equations (Rempel 2017 ApJ 834, 10) are solved with hydrodynamic or internal energy instead of total energy. Boris simplification of semirelativistic MHD equations can be solved by setting 'mhd_boris_simplification=T' and 'mhd_semirelativistic=F' to get faster but less accurate solutions. 'mhd_boris_simplification=T' is working with all versions of MHD equations including 'mhd_internal_e=T' and 'mhd_hydrodynamic_e=T'. Since semirelativistic MHD waves are very complicated, only approximate fast magnetosonic wave speed is implemented to use HLL or tvdlf scheme, schemes (such as HLLC and HLLD) depending on more wave speeds are not yet fully compatible with semirelativistic MHD. Note that when using semirelativistic MHD, the definitions of momentum and total energy are different from MHD, so call 'mhd_to_primitive' to get velocity and gas pressure from momentum and total energy instead of using the classic relations.
Synthetic EUV/SXR/whitelight emission
User can synthesize EUV images, EUV spectra, SXR images and whitelight images using 3D .dat files inside amrvac. These can be finished easily by adding some parameters into .par file. The images/spectra will be output to .vtu/.vti files when the convert_type in Filelist is set to 'EIvtiCCmpi', 'ESvtiCCmpi', 'SIvtiCCmpi' ... Thehe size of a pixel is the same as that in relevant observation data (such as SDO and RHESSI). The point spread function (PSF, instrument effect) has been included for 'instrument' resolution.
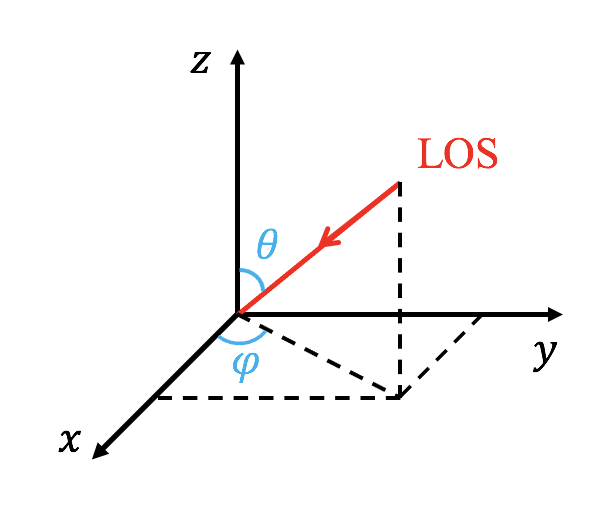
The line of sight (LOS) is controlled with 'LOS_theta' and 'LOS_phi', where the LOS is anti-parallel to the vector [cos(LOS_phi)*sin(LOS_theta), sin(LOS_phi)*sin(LOS_theta),cos(LOS_theta)] (see the following figure). The units of 'LOS_theta' and 'LOS_phi' are degree. User can rotate the image with 'image_rotate' (in degree). In cartesian coordinate, the y direction of the image is located in a plane given by the LOS and the z direction of the simulation data. In spherical coordinate, the definations of 'LOS_theta' and 'LOS_phi' are the same with theta and phi of the coodinate, but 'LOS_theta' and 'LOS_phi' are in degree.
The whitelight emission supposed observed by LASCO can be synthesized, and the instrument related parameter refers to 'whitelight_instrument'. User can specify the radius of the occultor of the instrument with 'R_occultor' (in solar radius).
The wavelength of an EUV image is defined with 'wavelength'. The bottom/upper cutoff energy of SXR image is defined with 'emin_sxr'/'emax_sxr' (in keV).
The wavelength of the EUV spectra is defined with 'spectrum_wl'. When 'spectrum_euv' is 'true', spectra at a slit in the corresponding image will be given. The output is a 2D image, where x-axis is wavelength and y-axis is space (physics distance at the slit). The slit is parallel to the y axis of corresponding EUV image (controlled by 'LOS_theta', 'LOS_phi' and 'image_rotate'). The location of the slit 'location_slit' is the x value of the image. The location of the slit 'location_slit' is the coordinate value at the third direction (perpendicular to LOS and slit). For example, the LOS along x direction and the slit along y direction, then 'location_slit' should be z of the slit. The domain in wavelength is controlled by 'spectrum_window_min' and 'spectrum_window_max'.
In cartesian, the mapping between simulation box coordinate and synthesized image coordinate is not only controlled by LOS, but also controlled by the parameter 'x_origin'. The simulation box point given by 'x_origin' will always located at (X=0,Y=0) of the synthesized image. The parameter 'big_image' is added for making movie that the LOS is changing. When 'big_image=T', then the synthesized EUV/SXR image will have a fixed big domain. The domain will not change when changing the LOS, and the whole simulation box will always be convered inside the domain. The parameter 'x_origin' is also added for making such kind of movie.
Two types of resolution are supported: data resolution and instrument resolution. In data resolution, the size of the image pixel is the same as the size of the finest cell. In instrument resolution, the size of a pixel is the same as that in relevant observation data (such as SDO and RHESSI). The point spread function (PSF, instrument effect) has been included for instrument resolution. The spatial resolution or EUV image/SXR image/EUV spectra is controlled by the parameter 'dat_resolution'. The default resolution is instrument resolution, and the data resolution can be activated by setting 'dat_resolution' to 'True'. Under the instrument resolution, the spatial resolution can be artificially changed with the parameter 'instrument_resolution_factor'. For example, we can double the spatial resolution by setting 'instrument_resolution_factor=2'.
The default unit of length for the outputed image is arcsec and the related variables in .par file, but user can set 'activate_unit_arcsec' to F to use the simulation length unit.
Only Cartesian and spherical coordinate system are supported currently. In spherical coordinate, region below 'R_opt_thick' (in solar radius, default value is 1) is assumed as not transparent.
&emissionlist filename_euv= CHARACTER wavelength= 94 | 131 | 171 | 193 | 211 | 304 | 335 | 1354 | 263 | 264 | 192 | 255 LOS_theta= DOUBLE LOS_phi= DOUBLE image_rotate= DOUBLE x_origin(1:3)= DOUBLE big_image= LOGICAL filename_sxr= CHARACTER emin_sxr= INTEGER emax_sxr= INTEGER filename_spectrum= CHARACTER spectrum_wl= 1354 | 263 | 264| 192 | 255 spectrum_window_min= DOUBLE spectrum_window_max= DOUBLE location_slit= DOUBLE filename_whitelight= CHARACTER whitelight_instrument= 'LASCO/C1' | 'LASCO/C2' | 'LASCO/C3' R_occultor= DOUBLE R_opt_thick= DOUBLE activate_unit_arcsec= LOGICAL dat_resolution=LOGICAL instrument_resolution_factor=INTEGER /
Splitting of the hydrostatic equilibrium
set
to true in mhd_list and implement usr_set_equi_vars in mod_ust.t file